
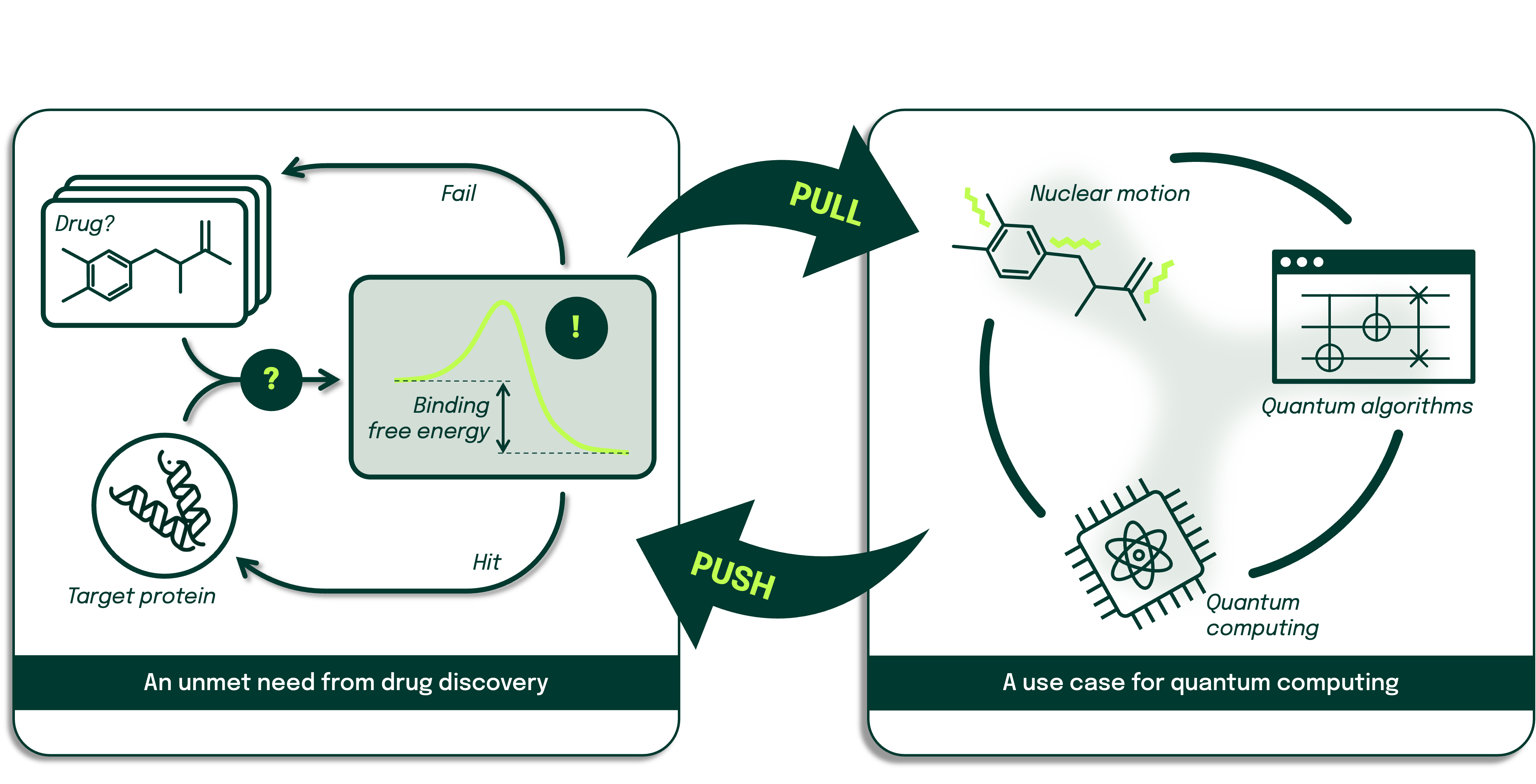
The challenge of accurate binding free energy
In the complex world of drug discovery, one crucial challenge stands out: determining how strongly a potential drug molecule binds to its target protein. This binding strength, often measured as binding free energy (ΔG), is a key indicator of a drug's effectiveness. While experimental methods can determine ΔG, they're often expensive and time-consuming, pushing researchers towards compute-intense and often inaccurate computational alternatives.
Limitations of traditional computational methods
Traditional computational approaches, whether powered by artificial intelligence or physics-based models, offer faster and more cost-effective solutions. However, they often face a significant limitation: the oversimplification or complete omission of entropic contributions. This simplification, while making computations more manageable, can lead to less accurate ΔG estimates.
Inaccurate ΔG estimates may compromise the reliability of predicted drug candidates for which the cost implications can be substantial. A false positive can lead to millions of dollars spent synthesizing and testing ineffective compounds, while a false negative might mean overlooking a promising drug candidate. In the pharmaceutical industry, where the average cost of bringing a drug to market exceeds one billion dollars and takes 10-15years, such computational inaccuracies can significantly impact both development timelines and budget.
The advantage of quantum computing
Quantum computing emerges as a promising solution to this challenge. It offers the potential to relax assumptions and simplifications that current computational methods employ to trade accuracy for speed and may therefore deliver ΔG predictions with enhanced accuracy. This in turn reduces uncertainties in drug candidate rankings and could with further maturation of quantum computers bring computational methods on par with experimental methods in terms of reliability of predicted binding energies. In the longer term, quantum computing may enable us to handle complex molecules that are intractable for classical computers.
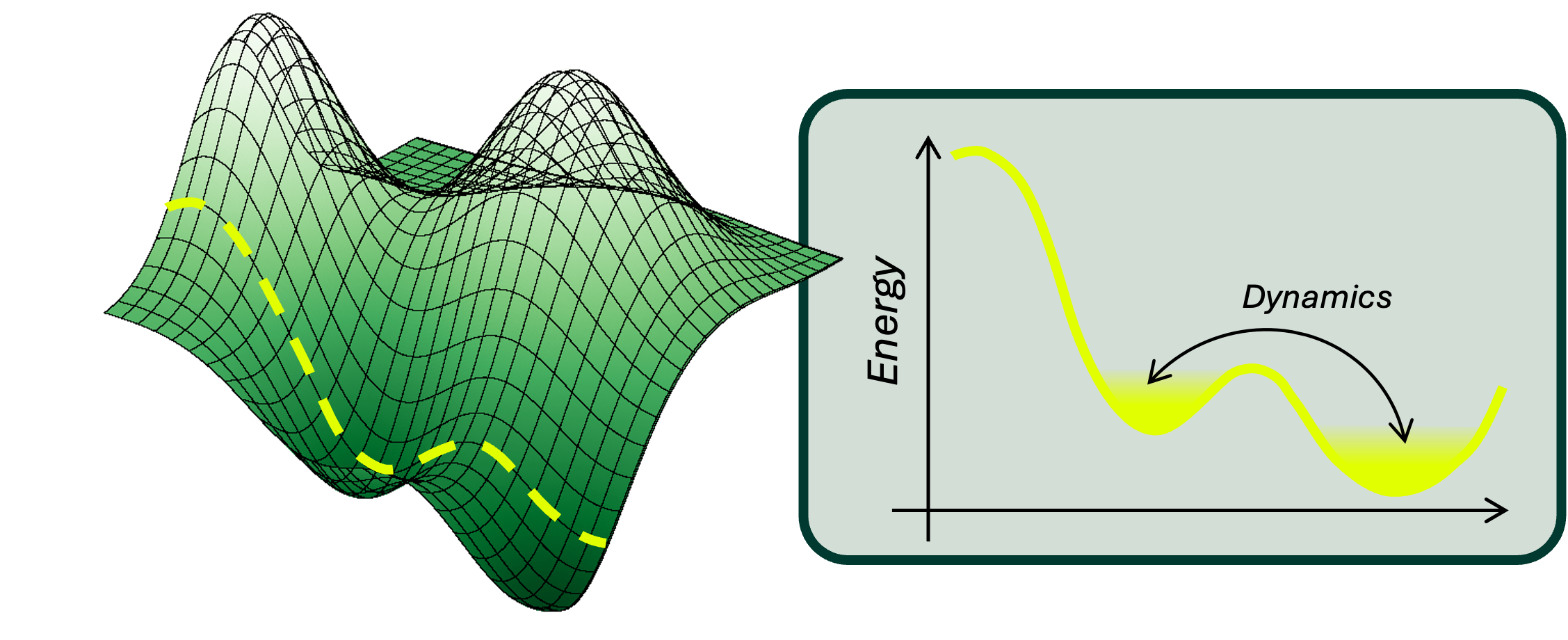
Vibrational energies and the entropic contribution
Vibrational energies directly contribute to the entropic components of ΔG through molecular motion. At the molecular level, atoms are not static — they constantly vibrate around their equilibrium positions, and these vibrations correspond to quantized energy levels. The collective set of these vibrational modes determines how a molecule stores and exchanges thermal energy. Because entropy is fundamentally tied to the number of accessible microstates a system can explore, the distribution and nature of vibrational energy levels directly influence the entropic component of ΔG. Accurately capturing these vibrations, especially low-frequency modes associated with large-scale molecular motions, is essential for predicting how tightly a ligand binds to a protein.
While current computational methods often rely on harmonic approximations of molecular vibrations to simplify calculations, these approximations break down for the anharmonic potentials typical in drug-protein interactions, where molecular motions can be large and complex. This limitation is particularly severe when considering thermal effects, which require accurate treatment of anharmonicity for reliable ΔG predictions.
Our quantum approach to anharmonic vibrational calculations
To address the challenges of accurately modeling complex molecular vibrations, especially those that go beyond harmonic assumptions, our team has developed new methods to perform these calculations on fault-tolerant quantum computers exploiting qubitization and the Quantum Phase Estimation [Majland et al. (2025)]. Our innovations include implementation of realistic potential energy surfaces which capture complex anharmonic effects which are beyond the harmonic approximations. This helps us better predict how molecules actually move and interact, leading to more accurate calculations of their energies. In addition, we implement low rank decompositions of the potential energy surfaces which simplify the complex interactions between different vibrations by breaking them down into smaller, easier-to-handle parts. We also implement parallelization techniques which splits calculations into many smaller tasks that can be run at the same time.This approach drastically speeds up computations and cuts down the time and cost needed, making it possible to study larger and more complex molecular systems efficiently.
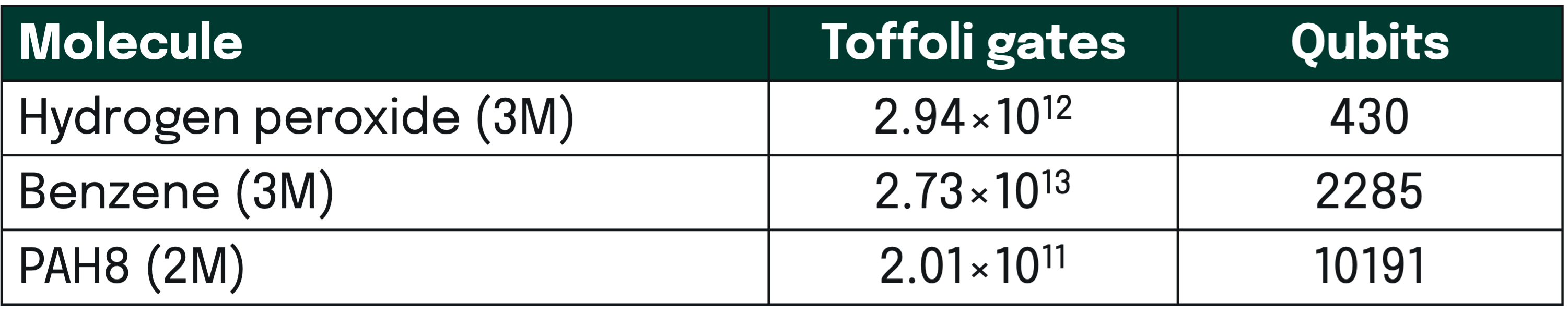
A central part of our work is to also estimate the ressources needed for simulating vibrational properties of molecules with fault-tolerant quantum computing using the developed techniques. Here, we provide just results from a few examples of molecules analysed to give an idea of the technological maturity needed. For the full set of results we refer to the scientific paper [Majland et al. (2025)].
A step toward more accurate drug discovery
While our current results represent an encouraging first step, more work is needed to fully compete with experimental methods in ΔG determination. Nevertheless, our research has established a foundation for more accurate vibrational energy calculations. Looking ahead, ongoing improvements in quantum hardware and algorithms may help increase accuracy and allow for more complex simulations of drug-protein interactions. Over time, this could help speed up parts of the drug development process, lower costs, and improve the chances of finding effective drug candidates. In the future, as quantum computers and their software get better, we expect to run more accurate and detailed simulations of how drugs interact with proteins. This could gradually make drug development faster, less expensive, and more reliable.
References
Majland et al. “Fault-tolerant quantum computations of vibrational wave functions”, https://arxiv.org/pdf/2508.16253(2025)



