Good, Better, BEAST!

Published on March 31, 2026
Kvantify Research and Applications
With Qrunch 1.1 we just notched-up state of the art on both accuracy and speed of chemistry simulations on quantum computers.
“How do I narrow down and develop the chemistry applications of quantum computing that can create value for me?” This is a hassle we often encounter among chemists and potential end users of the technology. Without a solid background in quantum computing, it’s hard to know what to expect from the technology or what to dream of, not to mention building the capability to actually test it out. With Qrunch we deliver the technology to bridge that gap.
The umbilical that feeds and enables the development of real-world and value-creating applications of quantum computing can be pictured as a three-strand braided cord of scale, speed, and accuracy, respectively. They are each necessary and highly interdependent, and striking the optimal trade-off is a fundamental challenge for discovering first applications with real-world impact.
When we launched Qrunch back in November 2025, we demonstrated that our technology allows scaling of chemistry computations to very large active spaces. We showcased the capability to exploit fully the computational power of Rigetti and IQM hardware to solve problems related to covalent ligand binding in drug discovery and electrolyte dissociation for battery technology, respectively. In the meantime, we have been busy braiding in substantial pieces of the accuracy and speed strands, extending and strengthening the cord significantly. We just made them available with the release of Qrunch 1.1
High-Accuracy Quantum Computations
The core promise of quantum computing for advancing chemistry simulations is one of accuracy. In molecular discovery for new medicines, more accurate simulations will translate into significantly faster development cycles, increased success rates, and R&D cost reductions. With high-accuracy quantum computing methods, chemists will be able to model effects such as dispersive interaction and strong correlations, which are essential in many biological processes but cannot be captured with current numerical techniques. This will not only impact molecular simulations in pharmaceuticals but a whole plethora of chemistry applications crucial for global health and sustainability, such as enzyme discovery and engineering for biosolutions development and emerging energy technologies.
In the following we illustrate the capabilities of the impressive accuracy improvements coming out with Qrunch 1.1. This has been done by re-running existing use-cases against an exact reference calculation and our new performance enhancing features. The new features deliver results showing up to 85% improvement in energy accuracy relative to a CASCI reference, 88% improvement on quantum computation time, and fast large-scale noisy simulations that accurately capture hardware performance. For cost reasons, the bulk of our performance tests are done mainly on our proprietary quantum simulator. In our experience, this approach is accurate, but for completeness we confirm it by doing validation runs on physical hardware. For these runs we measured wall time and by extrapolating the observed scaling, we find wall time advantage against an exact reference from around 14 qubits (i.e. wall time advantage – not accuracy advance). Details are provided in the sections below.
Introducing PT2
A crucial technology development step taken with Qrunch 1.1 is the augmentation of our scalable BEAST-VQE algorithm by a second-order perturbative correction (PT2). This is a one-shot correction applied after the last iteration, improving accuracy beyond the paired-electron approximation inherent to BEAST-VQE. The energy correction is derived from operator expectation values that can be calculated on quantum hardware, and PT2 is therefore by construction ready to harness future quantum hardware developments. To demonstrate the impact of this improvement we went back to the above-mentioned electrolyte dissociation showcase which, more specifically, concerns simulation of the dissociation energy of butyronitrile. As before, the reaction is modelled by stepwise displacement of the nitrogen atom of the nitrile group, however, this time we use our BEAST-VQE algorithm. Here we run the computations with the plain quantum algorithm, then with augmentation by orbital optimization (OO; already available in Qrunch 1.0), and with our new perturbative correction PT2, as well as with OO and PT2 in combination. Results are compared to a classical configuration interaction (CASCI) reference in Fig. 1 below.
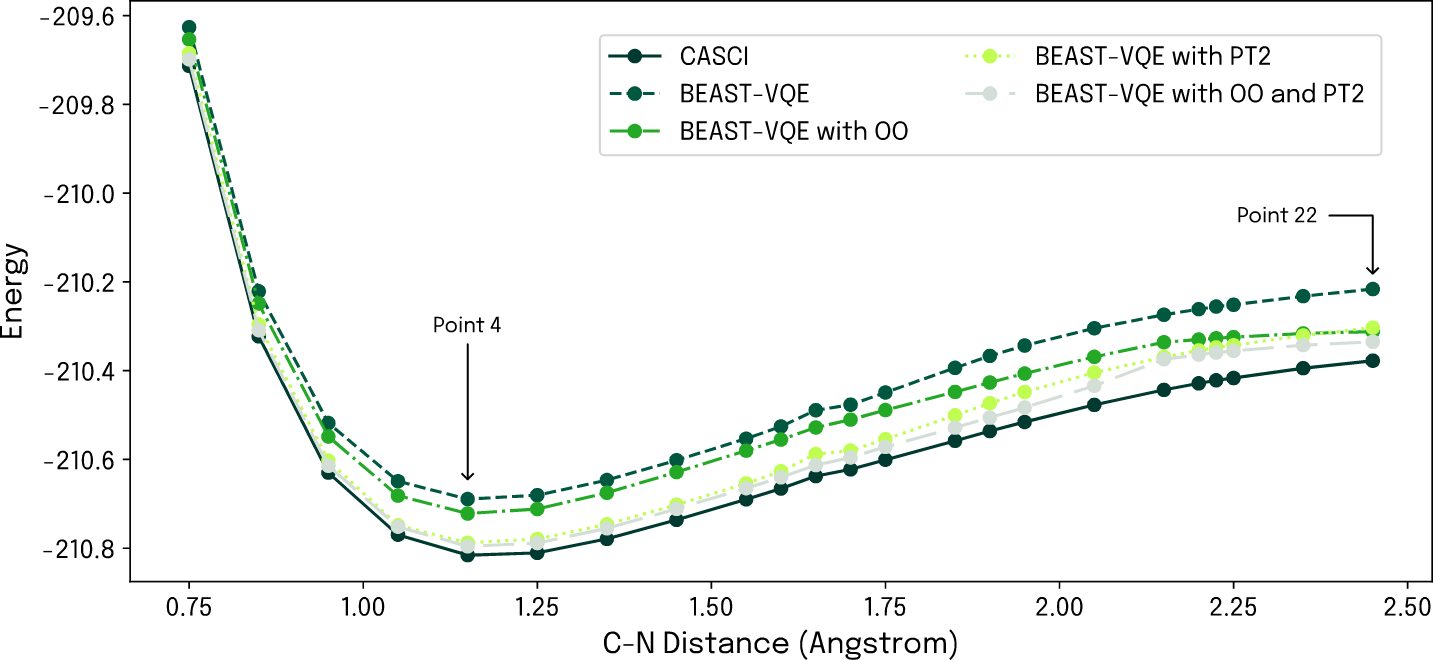
Figure 1: Simulated dissociation energy profile of butyronitrile. The impact on accuracy of augmenting BEAST-VQE by OO and PT2 is clear and the combination of both brings the quantum computation close to classical CASCI reference.
As a first assessment, we simulate the performance of the four computational approaches over the entire dissociation energy profile. Taking a step up from the previous sto3-g basis set, we now use 631g and define an active space of 14 spatial orbitals and 14 electrons. As evident from Fig. 1, the PT2 correction clearly shows its worth in comparison to our existing methodology and CASCI. It provides a significant accuracy improvement around the minimum of the energy profile though less so for larger atomic displacements. However, in this region OO offers a helping hand thanks to its roughly constant offset from the classical reference level. Crucially, the two augmentations are not mutually exclusive but can be combined to complement each other, as also shown in Fig. 1. An interesting observation is that the OO + PT2 augmented BEAST-VQE delivers results with an accuracy approaching closely the classical CASCI reference. Across the dissociation energy profile, we achieve an average error relative to CASCI of 0.033 ± 0.017, and compared to plain BEAST-VQE, the relative improvements are in the range of 59-86%.
The minimum of the energy profile at the equilibrium geometry (reaction point 4) and the fully dissociated configuration (reaction point 22) are points of particular interest and will be our focus for the following investigations. To assess the real-world impact of the new features, we supplement the simulation results with data from actual quantum hardware executions for these points. To this end, we use IQM’s 20-qubit QPU Garnet on IQM Resonance. We choose this backend simply because it fits well for the size of system we are interested in with these investigations. The results in Fig. 2 show how precision scales with system size, represented by the number of included spatial orbitals. We stop at 16 orbitals – beyond that point, our classical computations for establishing the CI reference run out of memory on developer class laptop.
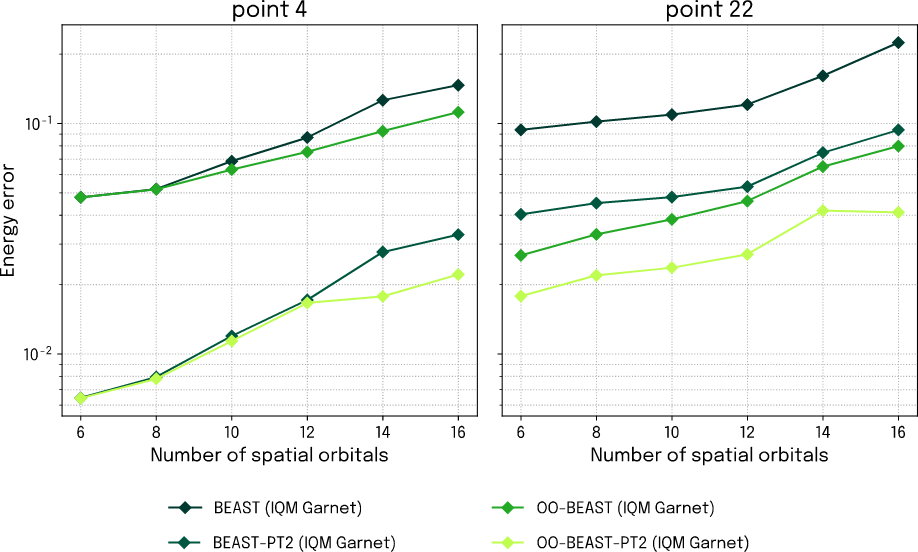
Figure 3: Energy error as a function of spatial orbital number for BEAST-VQE calculations at the equilibrium (left) and end point (right) of the dissociation energy profile executed on IQM Garnet.
The curves of the figure examine more closely the QPU performance at the identified points of interest, and we observe that the simulated performance and interplay between OO and PT2 is nicely reproduced on actual quantum hardware.
Fig. 1 clearly showed that the combination of OO and PT2 adds a consistent and significant improvement to BEAST-VQE, and the results from IQM Garnet in Fig. 2 verify the improved accuracy and further demonstrate that it is transferable to larger orbital spaces. The results thus pave the way for high-accuracy large-scale chemistry simulations on quantum computers. Furthermore, this shows that quantum computing can already provide high accuracy for active spaces that challenge classical CI methods, which are highly accurate but become computationally infeasible for problems beyond 16 qubits on a standard laptop.
Clocking the Accuracy
We now turn our attention to the speed-strand of the braid. Our FAST-VQE algorithm offers a convenient reference as it provides in-principle exact results if sufficient iteration steps are invested in the computation. Re-running the energy profile simulation in Fig. 1, we allowed FAST-VQE 100 iterations to obtain high accuracy, and completing the full reaction path took more than 3 hours. In comparison, the BEAST-VQE with OO achieved its limiting accuracy in only 25 iterations after which the PT2 correction was applied on top, completing the job in less than 20 minutes. That’s an 88% speed-up – at the price of only a small compromise on accuracy.
Going deeper into the matter, we first zoom in on the equilibrium and end points of the dissociation reaction again, only now we clock the total computation time for simulated BEAST-VQE as the number of spatial orbitals is scaled (Fig. 3). An immediate and very important observation is that the classical (CASCI) and quantum simulations scale very differently, with the classical one taking off along a much faster exponential than the simulated quantum computation. This explains why our CI reference is limited to 16 spatial orbitals and really underlines the potential of porting molecular simulations to quantum computers.
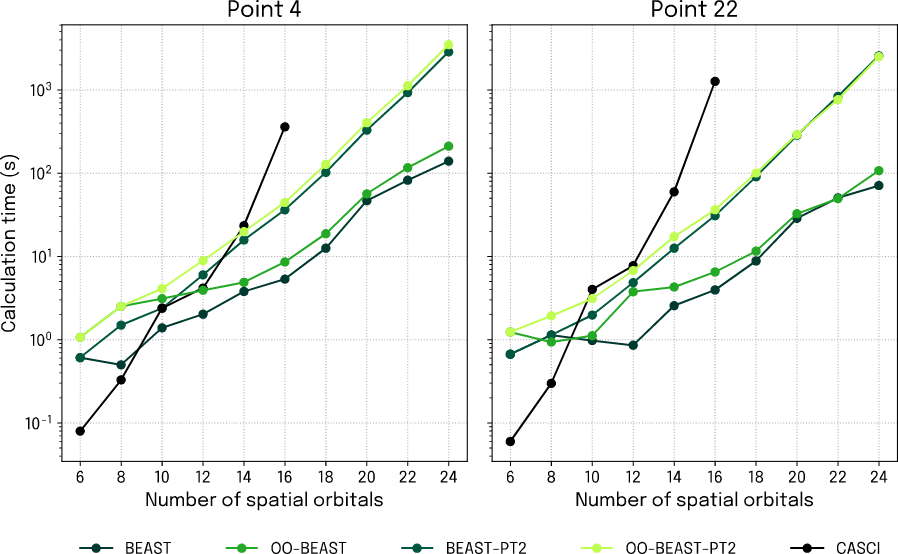
Figure 5: Calculation time as a function of spatial orbital number for simulated BEAST-VQE calculations at the equilibrium (left) and end point (right) of the dissociation energy profile.
Next, we bring all three strands together and establish a holistic view on the complete braid of scale, speed, and accuracy, considering both simulated and QPU executed performance. In Fig. 4 we plot the attained accuracy against the calculation time for BEAST-VQE and each of the three augmentations considered. In each case, we scale the computation from 6 to 16 spatial orbitals, and the accuracy is evaluated as the resulting energy error averaged over each of the 6 scaling steps. The lower panel provides the calculation time for the corresponding CASCI reference computations.
Looking first at the simulation results (circular markers) a clear accuracy hierarchy is established between the BEAST-VQE augmentations with both PT2-enabled methods coming out superior. This confirms the hypothesis that led us to develop PT2 in the first place, namely that the approximate nature of BEAST-VQE can to a large degree be corrected for while maintaining the scalability and speed of the algorithm.
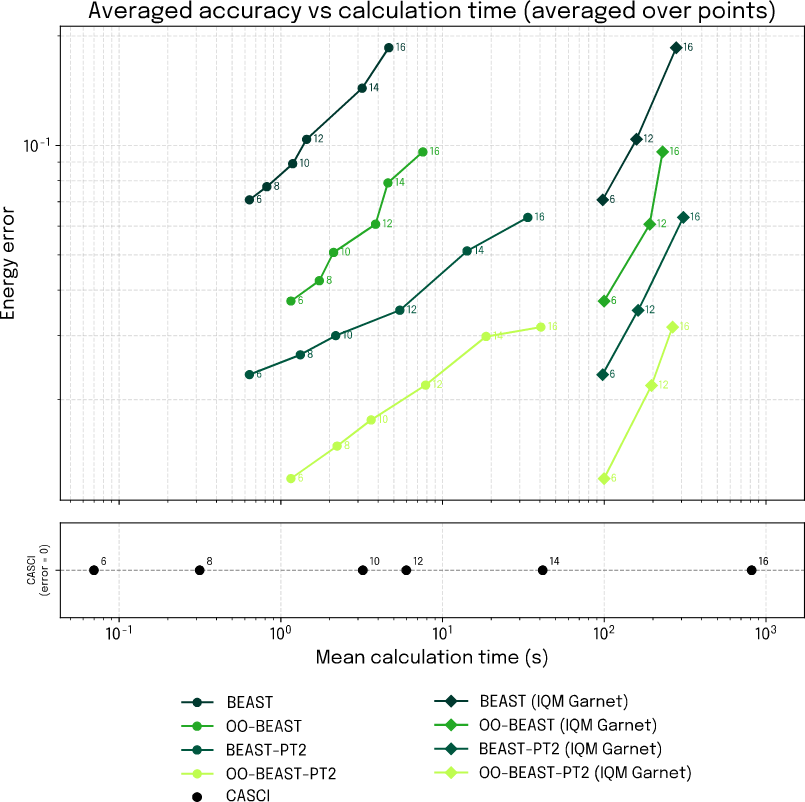
Figure 6: Attainable accuracy for simulated and hardware performance as function of calculation time and for increasing number of included spatial orbitals.
Turning next to the results obtained with IQM Garnet (diamond markers), it is important to keep in mind how public cloud quantum computers work to properly understand the figure. The only timing we have available for measurement is the total time consumption, including queuing time, transpilation time, transpiler switching etc. Those factors can make it difficult to get a clear picture of how the calculation time scales with problem size. Nonetheless, we observe that all of our QPU timings lie below the wall time of the 16 orbital CASCI calculation, clearly demonstrating a time advantage from quantum computers from around 16-qubits and up. Extrapolating the results indicate that a large advantage on calculation time stands to be gained for large qubit-count computations. However, the above considerations warrant that further investigations are conducted to firmly establish the QPU wall time scaling. Looking at the accuracy, we do, however, clearly see that the aforementioned hierarchy carries over to the QPU. Importantly, the IQM Garnet results indicate that the calculation time scales at a much more favorable rate than the CASCI reference while maintaining high accuracy. With our augmented BEAST-VQE algorithm, quantum computers already now offer a compelling way to access active spaces that are out of bounds with numerically exact classical methods and the gap will grow exponentially as QPU qubit counts climb.
GPU-Accelerated Noisy Simulations for Application Development
As we have just seen, fast and accurate simulation of quantum computations is instrumental for assessing algorithmic performance. The ability to realistically emulate quantum hardware is an equally important aspect of developing and scoping quantum computing applications and identifying high-value opportunities. To that end, Qrunch users can benefit largely from new features added in the 1.1 release.
Complementing our existing efficient memory restricted simulator, Qrunch 1.1 adds Monte Carlo sampling for noisy simulations to emulate hardware-like behavior based on calibration data. This allows users to quickly swap between any hardware backend supported by Qrunch and, based on latest calibration data available from the provider, investigate device performance and viability of running applications on actual hardware. Supporting qubit counts beyond what is accessible through traditional methods, our noisy simulator offers a powerful tool for development and testing of applications at practically relevant scale.
We have brought our noisy simulator to the test, and the results are very convincing (Fig. 5). Running 40 iterations of BEAST-VQE on IQM’s 54-qubits Emerald processor, we demonstrate very clear correspondence between actual hardware performance and the results from our noisy simulator. The simulation faithfully captures at what point in the computation the impact of hardware noise causes the results to deviate from exact simulation of noiseless performance.
Integrating such noisy simulations into an application development workflow will provide clear guidance on what to expect from the hardware, and it will not add any significant time overhead. Using a standard laptop, the 54 qubit simulations (all 40 iterations) plotted in Fig. 5 each completed in just 4.5 minutes. Out of that, less than half was spent on the noisy simulation part.
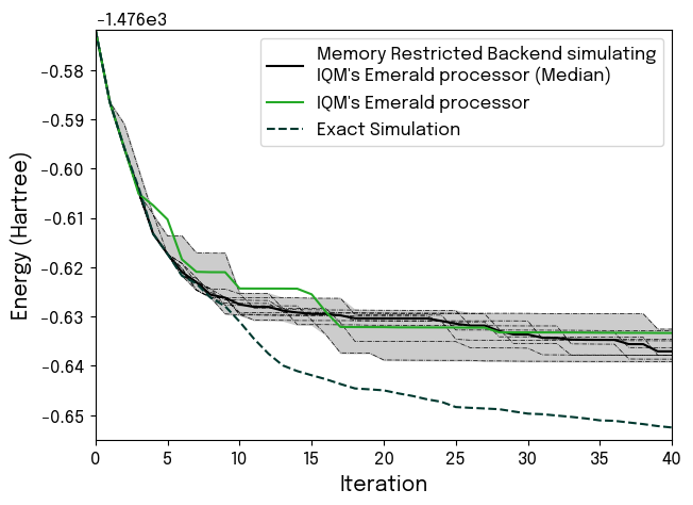
Figure 7: Comparison of the convergence of 54-qubit computation as simulated with Kvantify’s noisy simulator and executed on IQM’s Emerald QPU. The deflection point where the impact of noise causes the hardware performance to separate from the exact case is convincingly reproduced by the noisy simulation.
Exploiting GPU architectures is an obvious way to supercharge and gain further scale on quantum computing simulations through tensor network-based methods. With Qrunch 1.1 we have introduced a new GPU acceleration module providing improved performance for selected workflows.
GPU-accelerated simulations open up very exciting opportunities for chemistry and we have other ongoing developments in that area. More details on this work can be found in our recent blog post: https://www.kvantify.com/blog/nvidia-dgx-spark.
Charting the Landscape of Chemistry Applications
In analogy with how Harrison’s marine chronometer provided explorers with the needed tool for navigating the oceans and charting the world, we provide quantum explorers with the accuracy and simulation capability to explore and assess otherwise inaccessible regions in the landscape of chemistry quantum computations. The availability of large-scale noisy simulations based on latest hardware calibrations is a very powerful resource for exactly that. Equipped with a laptop alone, chemists can explore applications in fast development cycles and at the scale of modern quantum computing devices. Configurational variations of identified problems can be correlated with simulator performance to learn what makes the corresponding quantum circuits hard or unfeasible to simulate classically. With those landmarks at hand, roadmaps for how and when particular chemistry applications will benefit from quantum computing can be created, providing clear guidance for adoption of the technology.
We developed Qrunch to make quantum computing accessible to chemists and turn them into quantum explorers. They have the domain knowledge and experience to tell where the existing classical methods struggle or fall short, and with Qrunch we enable them to actively participate in charting the landscape of applications for quantum computing. With Qrunch 1.1 we have gone further and now provide users with a much stronger platform for orchestrating their exploration and digging deeper with respect to scale, accuracy, and speed.
Our technology has the capability to go well beyond 100-qubits chemistry computations, and it is top of our list to demonstrate this on quantum hardware. As we have shown here, a wall time advantage is already present, and the next crucial milestone for chemistry applications is to combine scale with an accuracy advantage. Towards that, demonstration of accuracy on par with density functional theory and capturing of interaction details intractable with current methodologies are high-priority targets that should also warrant attention from drug discovery and life sciences in general.