Qrunch
Showcases
Explore how Qrunch enables breakthrough quantum computing applications across renewable energy, biotechnology, and climate tech. Our showcases demonstrate real-world implementations on current quantum hardware, pushing the boundaries of what's possible today.
Simulating covalent ligand binding
drug discovery
covalent drug
Motivation
Covalent ligands are a fascinating and powerful class of drugs that bind to their biological targets by forming a covalent bond, well-known examples being aspirin and penicillin. This type of binding offers several compelling advantages: enhanced selectivity, prolonged duration of action, and improved potency compared to non-covalent drugs. However, this power comes with a challenge: such strong interactions also run the risk of off-target effects and unintended chemical reactions, which can lead to adverse outcomes.
From a computational perspective, modeling covalent binding is a difficult task. Most conventional tools, such as force fields, often fall short when it comes to simulating bond formation or breaking. This limitation arises because these empirical approaches rely on pre-fitted parameters derived from existing molecular configurations, making them poorly suited for capturing the complex quantum mechanical changes that occur during chemical transformations where electron distributions fundamentally shift, and new bonding patterns emerge. Quantum chemistry methods can overcome these limitations and provide crucial insight into drug-binding mechanisms by directly modeling the quantum mechanical foundations of molecular interactions. Quantum computing, in principle, may unlock the computational power needed to scale quantum chemistry simulations, allowing researchers to model intricate binding mechanisms and molecular transformations with enhanced accuracy and detail that drug discovery demands.
The case
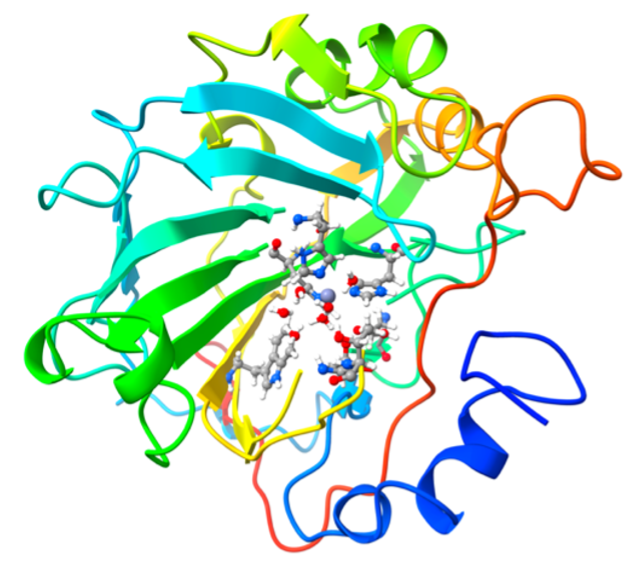
The Cathepsin-K enzyme is a protease involved in breaking down bone and cartilage and is actively studied as a therapeutic target for postmenopausal osteoporosis. The odanacatib ligand is a potent and highly selective covalent inhibitor of Cathepsin-K.
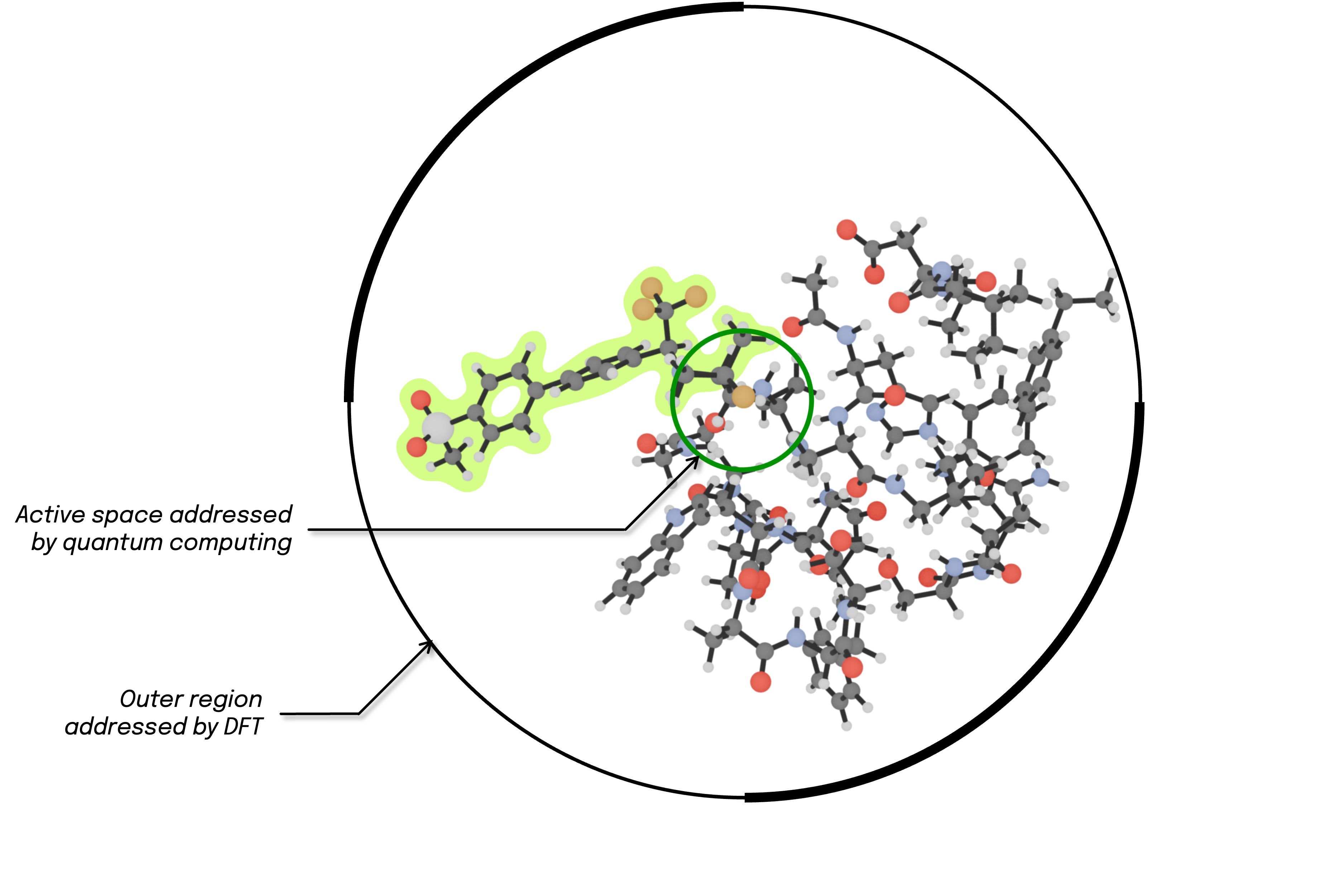
To set up the computation, we assign orbitals to the active space after a localization procedure that uses the electrophilic carbon from the ligand's nitrile functional group and the nucleophilic sulfur atom from the Cys-25 Cysteine residue in Cathepsin-K as defining centers for the quantum computing region. The surrounding environment of these active orbitals is modelled with projective embedding, where Kohn-Sham Density Functional Theory (DFT) provides the methodology to incorporate environment effects into the core active region. The pool of all paired excitations will then be generated from this active space and will be added via the gate selection procedure of the BEAST-VQE algorithm on a quantum device or, alternatively, on a simulator.
We executed the problem on different backends, going from state vector simulations to real quantum hardware. Using state vector simulations, we observe consistent and reliable energy differences between bound and unbound states across increasingly large active space definitions.
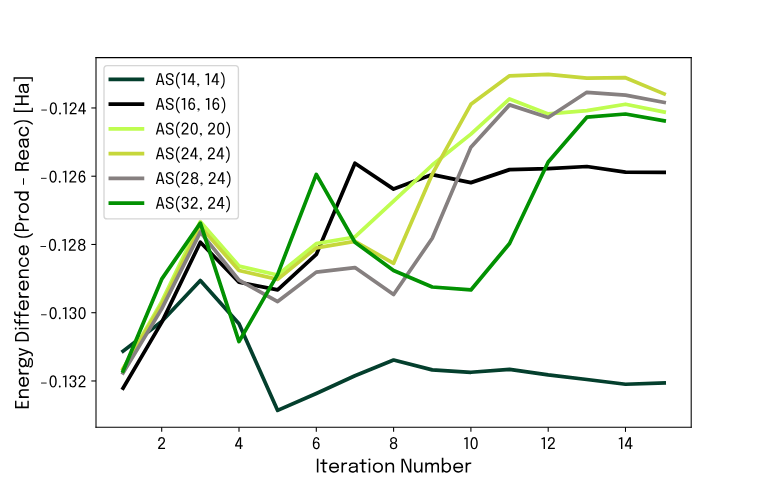
To keep the computation time feasible, going beyond 30 qubits resolution of the problem requires real quantum hardware, and for this task we used the Rigetti Ankaa-3 device, available via Amazon Braket. As explained above, VQE algorithms involve classical computations for certain tasks, which were executed with our exact chemistry-optimized simulator. As we scaled the problem to larger qubit numbers, this task was found to be a bottleneck — the classical computational workload increases dramatically as the size of the state vector grows. To address this challenge and enable full exploitation of Ankaa-3, Kvantify's memory-restricted simulator was deployed. It retains a user-specified number of state vector components, limiting memory usage, and enables simulation of systems of up to hundreds of qubits. Although this introduces an approximation, the impact on the final energy accuracy is minimal when a reasonable state vector pruning is chosen.
This enabled us to fully leverage the Ankaa-3 hardware to perform a calculation on 80 qubits, representing an active space of AS(80,24). The very encouraging observation is that the calculation performed on Ankaa-3 closely follows the state vector simulation, demonstrating the algorithm's scalability and noise resilience.

Key Results
The results are exciting because they point toward the future of quantum-enhanced drug discovery. BEAST-VQE cuts the number of required qubits in half, making it possible to simulate larger molecular systems on today's quantum hardware. It also simplifies quantum circuits and keeps the number of measurements constant — both key factors for making quantum simulations more efficient and scalable. This combined with further hardware developments in noise mitigation and scaling, is crucial for drug discovery, where understanding complex molecular interactions often pushes the limits of classical computing.
Conclusion
By improving the practicality of quantum simulations, our approach brings us closer to using quantum computers to accurately model drug candidates, speed up discovery, and reduce development costs. With these results, we have never been closer to the 100-qubit era of quantum chemistry simulations on real quantum hardware — a milestone that only a few years ago seemed far beyond reach.
Tutorial
A tutorial explaining how to set up the computations involved in this showcase is available here.
Toxicity prediction through bond dissociation energies
drug discovery
toxicity screening
drug design
Motivation
Bond dissociation energies (BDEs) are important in drug discovery for understanding the reactivity of molecules and quantifying how easily a specific bond will break to form free radicals, which in turn can lead to alkylation with consequent toxic implications. Mapping carbon-hydrogen (C-H) BDEs across a scaffold in ligands can inform drug design by identifying bonds prone to breaking, guiding safer substitutions, and prioritizing liabilities before costly in-vitro and in-vivo testing. Computationally accurate BDEs are required to capture subtle substituent effects essential for reliable predictions, and a correlated electronic-structure treatment beyond simple mean-field or empirical methods, is necessary.
Quantum computing offers a promising path forward as quantum algorithms for chemistry naturally encode electron correlations. Although current hardware is pre-mature for calculations directly useful in production drug discovery assessments, substantial improvements have been made in recent years both in terms of addressable system sizes and accuracy.
The case
To investigate this application of quantum computing and the feasibility of predicting BDEs with current quantum hardware, we explored modeling of BDEs for a selection of nitrosamines, characterized by different alkyl groups as substituents. Nitrosamines are toxic and can through metabolic activation become carcinogenic.

Although not directly focusing on C-H bond dissociation as typical for drug discovery applications, these molecules allow us to assess the effects of substituents in the stability of radical species following the N-N covalent dissociation.
The optimized structures were used as the onset for setting up classical CASCI reference calculations and FAST-VQE calculations with Qrunch. Appropriate care was taken to ensure consistent selection of orbitals for the equilibrium and dissociated states.
To establish classical references, mean-field baselines were established using unrestricted Hartree-Fock and Density Functional Theory, and subsequently CASCI correlated electronic structure calculations were performed. Moving on to computation of the correlated results on quantum hardware, an active space AS(10,10) was defined. This maps onto a 20-qubit computation which was executed on IQM Garnet. The deviation of the resulting energies from the CASCI reference for runs executed on quantum hardware as well as noise free state vector simulations obtained using Kvantify's proprietary chemistry-specific and efficient simulators included in Qrunch shows that, as expected, the deviation is larger for hardware runs subject to noise than for state vector simulations.

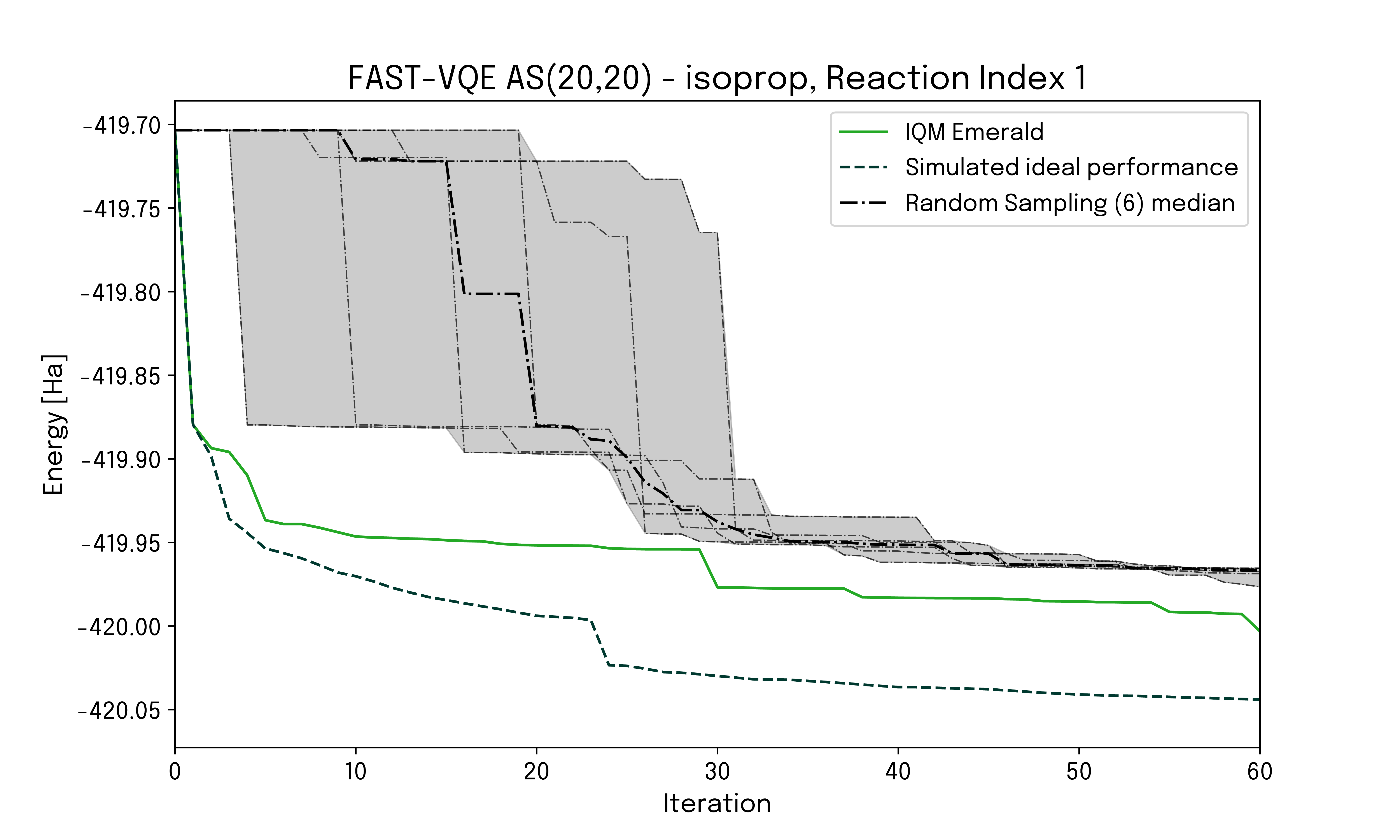
Scaling up the quantum computations further, calculations on an active space AS(20,20), mapping to 40 qubits, were executed on the IQM Emerald device. At this problem size, obtaining a CASCI reference is beyond our classical computing capabilities. To investigate the impact of hardware noise the energy evolution as function of the number of FAST-VQE iterations for hardware results are compared to a state vector simulation and simulated noise-dominated performance.
Key Results
BDE predictions obtained from quantum hardware calculations are in good correspondence with CASCI reference values. The simulated ideal curve exhibits a smooth and faster convergence without visible plateaus. This is expected as gate selection is not influenced by hardware noise. The energy convergence for the hardware run follows the ideal performance at the very beginning but gradually deviates towards a slower and flatter convergence trend, as a result of hardware noise. The noise-dominated curve exhibits a slower convergence, especially at the beginning where the correct selection of the initial gates leads to a steep decrease of the energy. As the number of iterations increases, noise effects make the hardware run approach the noise-dominated scenario.
Conclusion
We have shown how quantum computing can be effectively used as a promising computational tool for evaluating bond dissociation energies. Its value here is that quantum computing natively targets high-accuracy, correlated electronic structure, exactly what reliable BDEs modeling demands. This was probed for selected nitrosamines where the variation stems from alkyl substituents. In particular, the calculations predict lower BDEs for the substituted ones due to radical stabilization, as expected. The framework can be directly generalized to C-H bond breaking studies, highly relevant for toxicity prediction in drug discovery.
Carbon capture with COF-999
renewable energy
carbon capture
Motivation
Carbon capture is seen as a practical way to reduce CO₂ emissions from power plants and industries like steel and cement, where cutting emissions is especially difficult. While it's not a standalone solution, it could play a useful role alongside renewable energy and efficiency improvements in addressing climate change.
Carbon capture technology comes in many forms and shapes. While major point source capture technologies target capture at the origin of emissions, an emerging alternative is direct air capture that extracts CO₂ directly from the atmosphere using chemical or physical processes. To be efficient, used solid sorbents and liquid solvents must exhibit a high binding affinity for CO₂, but also be able to release it on demand for storage or reuse, preferably without degradation.
The case
A promising material for direct carbon capture is the porous COF-999, which shows enhanced CO₂ uptake under humid conditions. At the same time, the hydrophobic nature of the material allows low-energy regeneration, providing an efficient capture and release process.
The advantageous reaction and regeneration cycle of COF-999 is related to key transition states between adsorption and desorption. The goal for this showcase is to understand how the transition state energies of these CO₂ binding reactions in COF-999 depend on the presence or absence of water using a combination of classical and quantum computing.
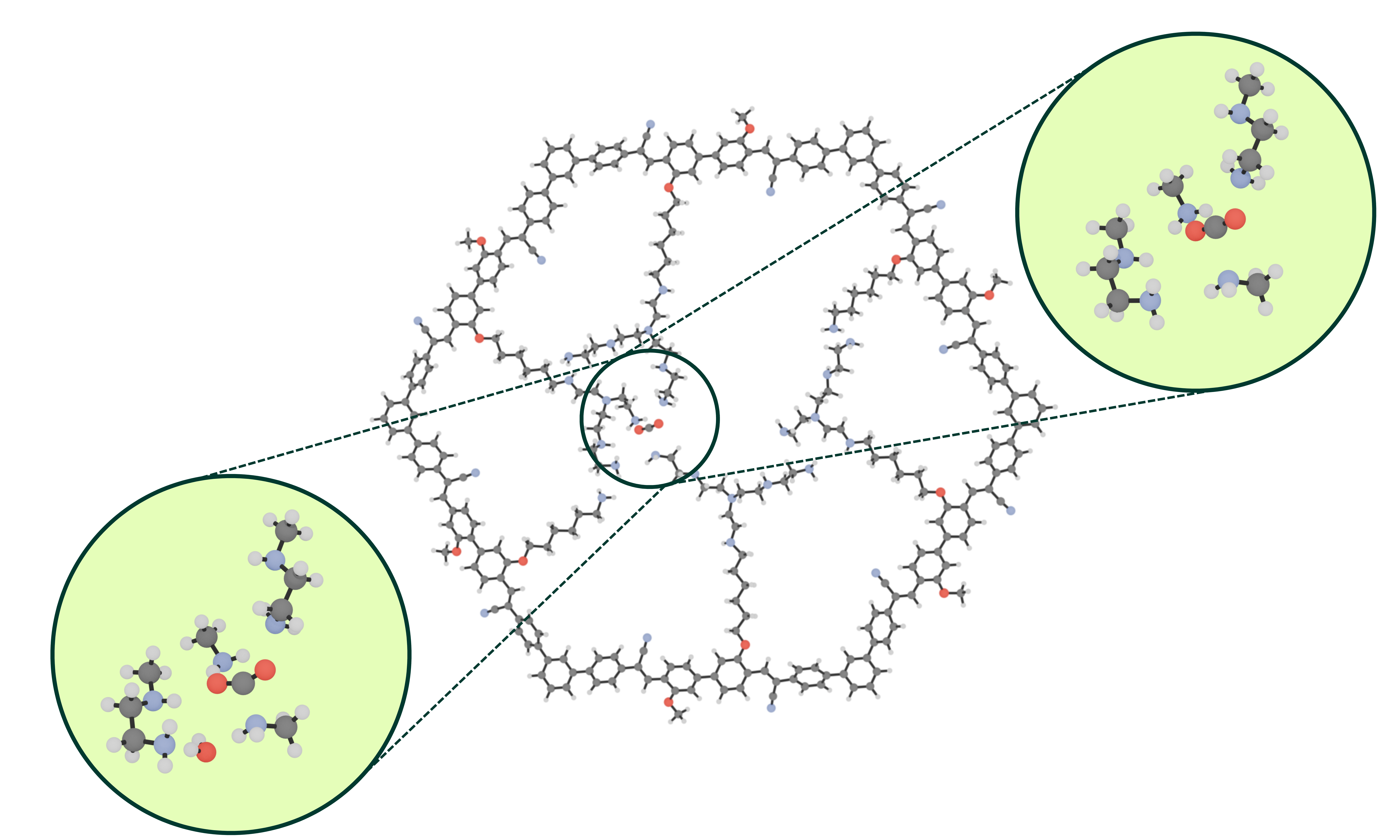
In COF-999, the reactive groups that capture CO₂ are amines (NH₂), and the capture happens through a transition state towards formation of carbamate. To investigate the influence of humidity, the transition state energies are calculated in the cases where a water molecule is present or absent in the proximity of the binding site to assess the impact on the activation energy barrier. In the presence of water, two possible reaction mechanisms exist: a direct proton transfer from the amine to the CO₂ or a proton exchange via a near-by water molecule.
To enable calculation of reaction energies and searching for transition states, reduced structures centered around identified starting and end points for the reaction were used in the two cases of interest. Reaction energies and transition states were obtained from a self-consistent tight-binding method (xTB). Using the transition state structure found by xTB, energies are calculated with a projective embedding method using Qrunch to run the FAST-VQE wave function method on a noise-free simulator for active spaces AS(10,10) and AS(24,24). In the former case, an exact reference energy could also be calculated from CASCI. The found reaction energies and transition state structures indeed show that the transition stage energies are found to be lowest in the presence of water.

To demonstrate the transferability of the problem to quantum computing, Qrunch was used to run FAST-VQE calculations on two different quantum hardware devices from IQM: Garnet with 20 qubits and Emerald with 54 qubits. In both cases the convergence of the algorithm as executed on actual hardware was compared to exact state vector calculations and a simulated noise-dominated hardware, allowing an empirical investigation of the hardware capability. Up to at least 10 iterations, the quantum hardware delivered better operator selection and in turn better convergence.

Key Results
It was found that accuracy could be improved moderately by increasing the shot count. Employing post-selection as a simple error mitigation strategy allowed a further improvement on convergence, comparing different strategies executed on IQM Emerald.

Conclusion
In this proof-of-concept we have demonstrated the feasibility of executing quantum chemistry calculations of transition state energies relevant to the understanding of carbon capture technology. Specifically, this was done for the example of COF-999 that shows promise for CO₂ capture, especially in humid conditions. This property was evidenced by how an additional water molecule in the proximity of the CO₂ binding site lowers the activation barrier for the formation of a covalent bond between CO₂ and COF-999. Using Qrunch, projective embedding calculations with FAST-VQE were executed on IQM quantum computers exploiting the full computational capacity of both the Garnet and Emerald chips, utilizing up to 48 qubits.
Computational modeling with quantum chemistry may help to guide the design of such materials by providing very accurate predictions of the performance without the need to synthesize and test the material first. In this way, many designs and material optimizations can be tested cheaply in silico before synthesis. Quantum computing holds the promise of radically speeding up very accurate simulations of such calculations.
Tutorial
A tutorial explaining how to set up the computations involved in this showcase is available here.
Enzyme engineering for bioremediation
bioremediation
enzyme engineering
Motivation
Bioremediation is an advanced and sustainable climate technology that exploits nature-based processes to mitigate and possibly completely eliminate the harmful effects caused by environmental pollutants in soil and water. It utilizes the metabolic activities of microorganisms and their enzymes to break down pollutants. One class of pollutants and toxins with significant environmental impact are haloalkane compounds that are widely used in industry as solvents, refrigerants, and propellants. Dehalogenase enzymes are biocatalysts that mediate the removal of halogen atoms from organic compounds by cleaving carbon-halogen bonds through the process of dehalogenation.
The case
A specific application of dehalogenase is for the conversion of 1,2-dicloroetane (DCE) which is a high-volume industrial chemical used in the production of PVC products and found as contaminant in drinking water. DCE is toxic, possibly carcinogenic, and a perennial pollutant. Conventional treatments are inefficient and expensive making biological bioremediation techniques an attractive alternative.
One possible approach is to use the dehalogenase enzyme to catalyze the conversion of DCE. The key reaction catalyzed is a nucleophilic attack, known as an SN2 attack,
in which the enzyme's active site attacks the carbon-halogen bond and displaces the halogen, in this case a chlorine atom. An intermediate covalent bond is formed with the carbon atom but subsequently hydrolyzed. Simulating such process may assist the development of highly performant enzymes for PFAS clean-up, but the required description of electronic correlations goes beyond mean-field approach. Quantum computing promises to enable such calculations and provide the accuracy required to support enzyme engineering.
In this showcase, we use Qrunch to set up a computation on IQM's quantum hardware using our proprietary algorithms FAST-VQE and BEAST-VQE. The goal is to provide an approximate ground state energy for the reaction described above. To exploit the available quantum computing resources maximally in the simulation, we focus on describing the most relevant part of the problem. Our model for the enzyme includes 86 atoms but using the projective embedding technique a subset of 5 atoms involved in the reaction are selected to constitute the embedded problem treated by quantum computing, while the remainder of the system is solved with Density Functional Theory. The electronic orbitals of the embedded region are described using an active space, AS(10,10), of 10 molecular orbitals and 10 electrons, mapping to 20 qubits computation.
Key Results
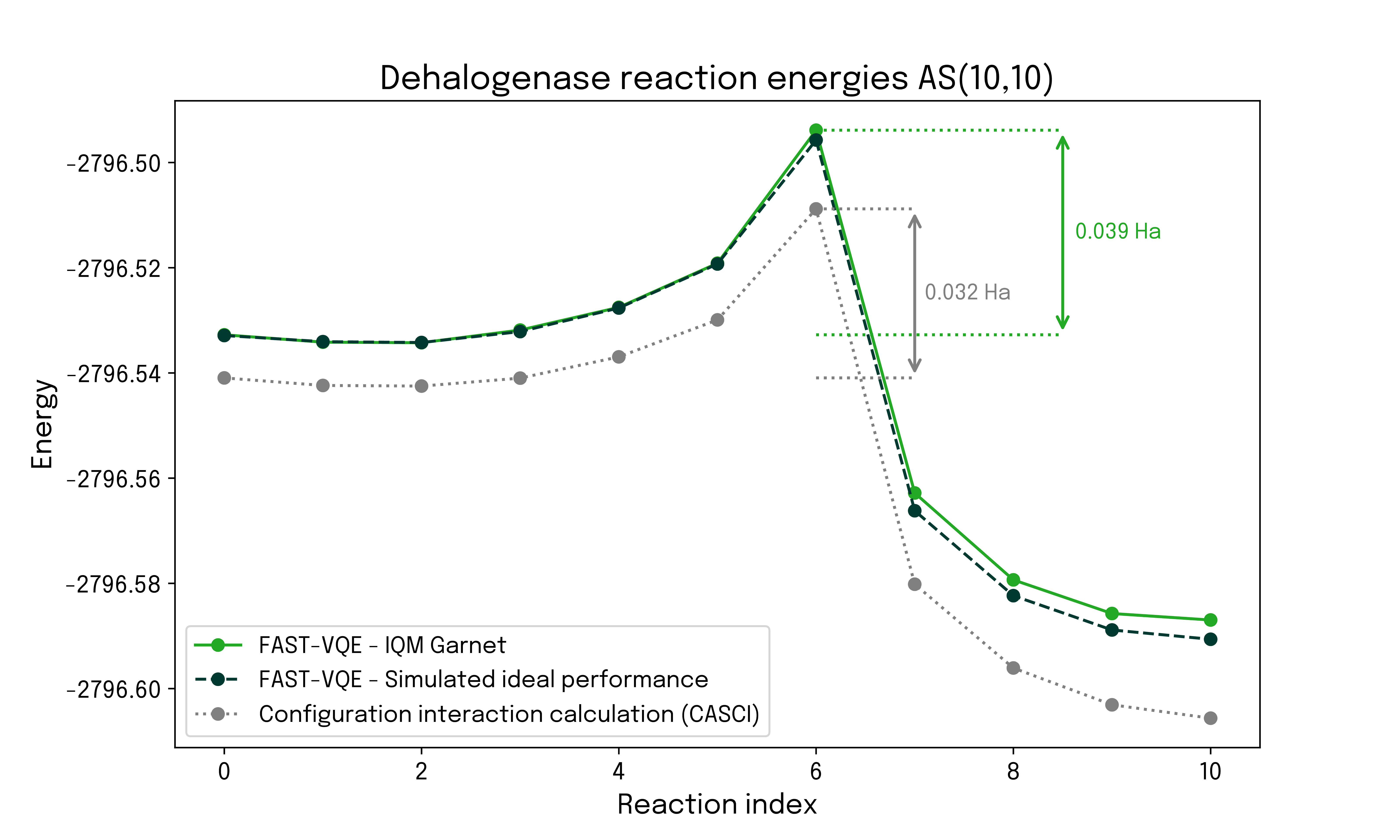
The calculated ground state energies for 11 points along the reaction path were calculated with FAST-VQE on IQM Garnet, exploiting the full 20 qubits of the device. In this regime we are still able to find the CASCI solution, which is included as a classical benchmark. A state vector simulation is also possible and provides a reference for the ideal performance of the hardware in the absence of noise.
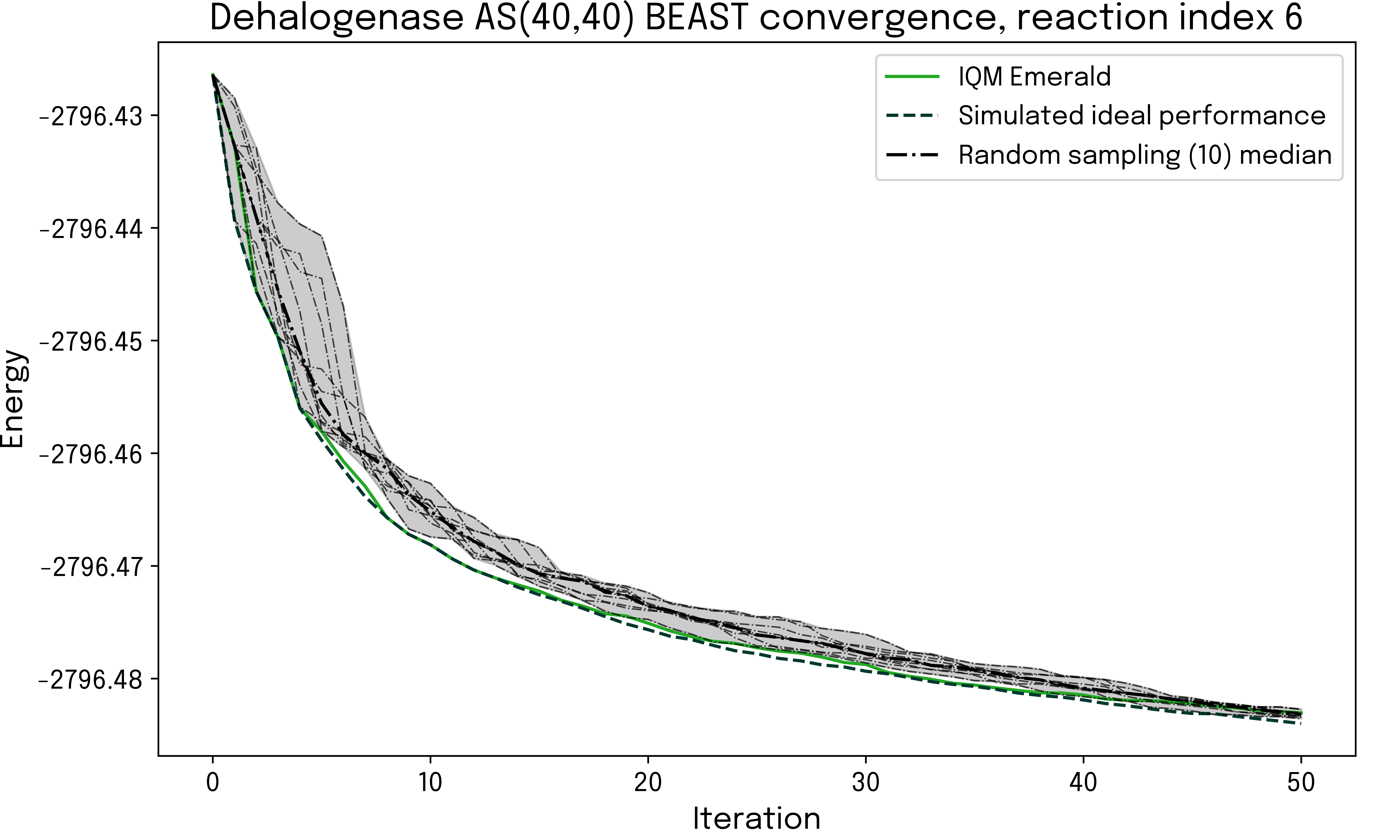
Next, we explore the scalability of the problem on IQM's hardware by going to an AS(40,40) active space of 40 spatial orbitals, which with the BEAST-VQE algorithm maps to a 40 qubits problem that can be executed on IQM Emerald. In this case, we are no longer able to establish the CASCI solution on classical computers. Focusing on a single reaction point we investigate the convergence of the BEAST-VQE ground state energy as function of iteration number and compare with two simulated hardware scenarios - ideal performance and noise-dominated performance. Strikingly, the results show that the actual quantum hardware performance is better than the noise-dominated case for most of the evolution of the system and closely aligns with the state vector simulation.
Conclusion
Enzyme-based bioremediation is an important climate technology for environment clean-up, and the computational complexity of simulating reactions to support development and enzyme engineering makes it a promising use case for quantum computing. We have successfully shown that with Qrunch, current quantum hardware can deliver reaction energies that closely match exact classical benchmark values. Scaling the problem to an active space mapping to 40 qubits computations with BEAST-VQE — beyond the point where we can establish the classical reference — we observe rapid and close to ideal convergence. Also, the accuracy of the calculated reaction ground state energies is determined by hardware.
Tutorial
A tutorial explaining how to set up the computations involved in this showcase is available here.
Enzyme engineering for catalysis
Enzyme engineering
nitrogen fixation
Motivation
Enzymes are vital for thousands of naturally occurring biological processes, acting as catalysts for biochemical reactions. The industrial application potential is vast, spanning fields as pharmaceuticals, food industry, chemicals, and climate tech. As enzyme activity has a high degree of specificity, detailed knowledge of the reaction process is needed to identify or engineer enzymes for target applications. However, enzymes are highly complex molecules, and processes cannot be modeled adequately with state-of-the-art classical computational tools. Thus, future industrial development in the field could be accelerated significantly by the development of novel computational techniques overcoming current limitations, making it a highly interesting use case for quantum computing.
The case
In this showcase, we demonstrate a near-term application of quantum computing in biotech by performing enzymatic reaction calculations on current quantum hardware. The specific challenge targeted is simulation of a reaction known as the proton transfer step for carbon dioxide hydration catalyzed by carbonic anhydrase. The enzyme carbonic anhydrase naturally catalyzes the conversion of carbon dioxide and water into carbonic acid, a process important for regulation of blood pH and transport of carbon dioxide in the body. Carbonic anhydrase is also used in climate tech for carbon capture and nitrogen fixation.
Density Functional Theory (DFT) is a well-established and widely used method for quantum mechanical modeling of electronic structures on classical computers. However, the approach struggles to reach chemical accuracy, an error level defined as 1 kcal/mol, required for quantitative modeling of chemical reactions. There is thus a strong incentive for developing methods that can augment DFT modeling of molecules in regions of high importance. As evident, enzymes are indeed complex molecules. However, a confined active site can be identified, constituting the atoms that directly take part in the chemical reaction the enzyme catalyzes. Subdividing the molecule into regions of decreasing importance for the reaction allows for constructing a multilayered embedding of the enzyme where computational resources are allocated according to accuracy requirements. Quantum computing is leveraged for treating the active site, vital for understanding the reaction process, with highest possible accuracy while the surrounding central region is modeled with DFT and the outer regions using classical methods. Appropriate care is taken to match the models at the layer boundaries. Building problems with projective embedding is an integrated functionality in Qrunch, readily available and easy to use. The result is a hybrid quantum-classical computational problem that combines calculations executed on quantum hardware and high-performance computers. In this specific case, Kvantify's proprietary quantum algorithm FAST-VQE was employed for a wavefunction embedding of the active site.

Key Results
The hydration reaction was simulated by identifying a reaction pathway from the initial reactants to the final products via intermediate structures described by an abstract collective reaction coordinate. The resulting simulated reaction energies along the reaction pathway clearly showed the ability of the hybrid quantum-classical approach to closely match the classical exact benchmark from CASCI. Equally important, the convergence plots demonstrate that the approach can reach an error level well within chemical accuracy of 1 kcal/mol for sufficient iterations of FAST-VQE. The quantum algorithm was executed on quantum computers of two different modalities — ion trap and super-conducting respectively. Current quantum hardware allows for only limited circuit depth posing a challenge for iterative algorithms. To enable the required number of iterations, gate recompilation techniques were developed to compile the quantum algorithm into fundamental gate operations that require the lowest possible number of gates in each execution.
Conclusion
Exploiting the advanced problem building functionalities of Qrunch, the showcase demonstrates the utility of current quantum computers for industry relevant problems within biotech and the ability to deliver high-accuracy results. This opens a pathway for surpassing current limitations in the field and potentially leading towards exact chemical simulation results which could accelerate the development and widespread use of energy-efficient and environment-friendly enzymatic processes in industry, climate tech, and domestic households.
Electrolytes for tomorrow's battery chemistry
renewable energy
battery chemistry
Motivation
Electrification is a key strategy to combat energy crisis and climate change, and converting to renewable energy sources and increasing the electric capacity from renewables is essential. Making the shift requires efficient technologies for harvesting renewable energy, such as solar cells, and high-capacity fast-charging battery technology to meet the challenges of increased demand, load on charging infrastructure, and ensuring grid stability.
Electrolytes are crucial for the performance of batteries, and high-potency candidates are in increasing demand. One such candidate that is actively researched in the context of dye-sensitized solar cells (DSSC) and lithium-ion batteries, is butyronitrile (C₄H₇N).
The case
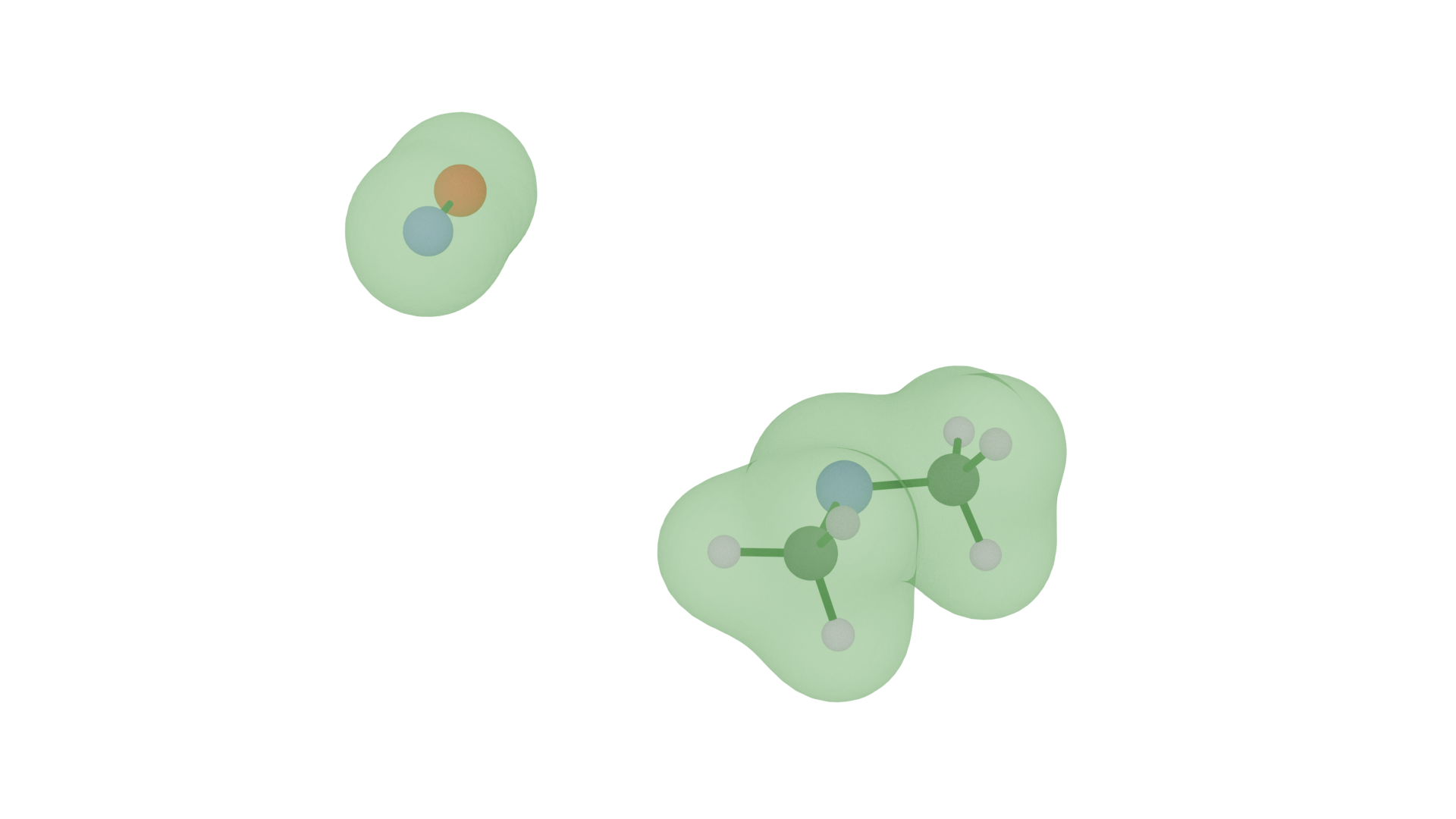
The dissociation of electrolytes is important for their functionality as medium for ion transport. While not relevant in that context, butyronitrile offers another dissociation process that is interesting seen from a quantum computing perspective, namely the dissociation of the nitrile group, which involves breaking of a triple bond between nitrogen and carbon. The bond of interest is that between the nitrogen and carbon atoms. The electron densities, derived from quantum-chemical calculations, show the light-green part of the electron density as representative of the effective calculation executed on quantum hardware while the dark-green part constitutes a static environment contribution.
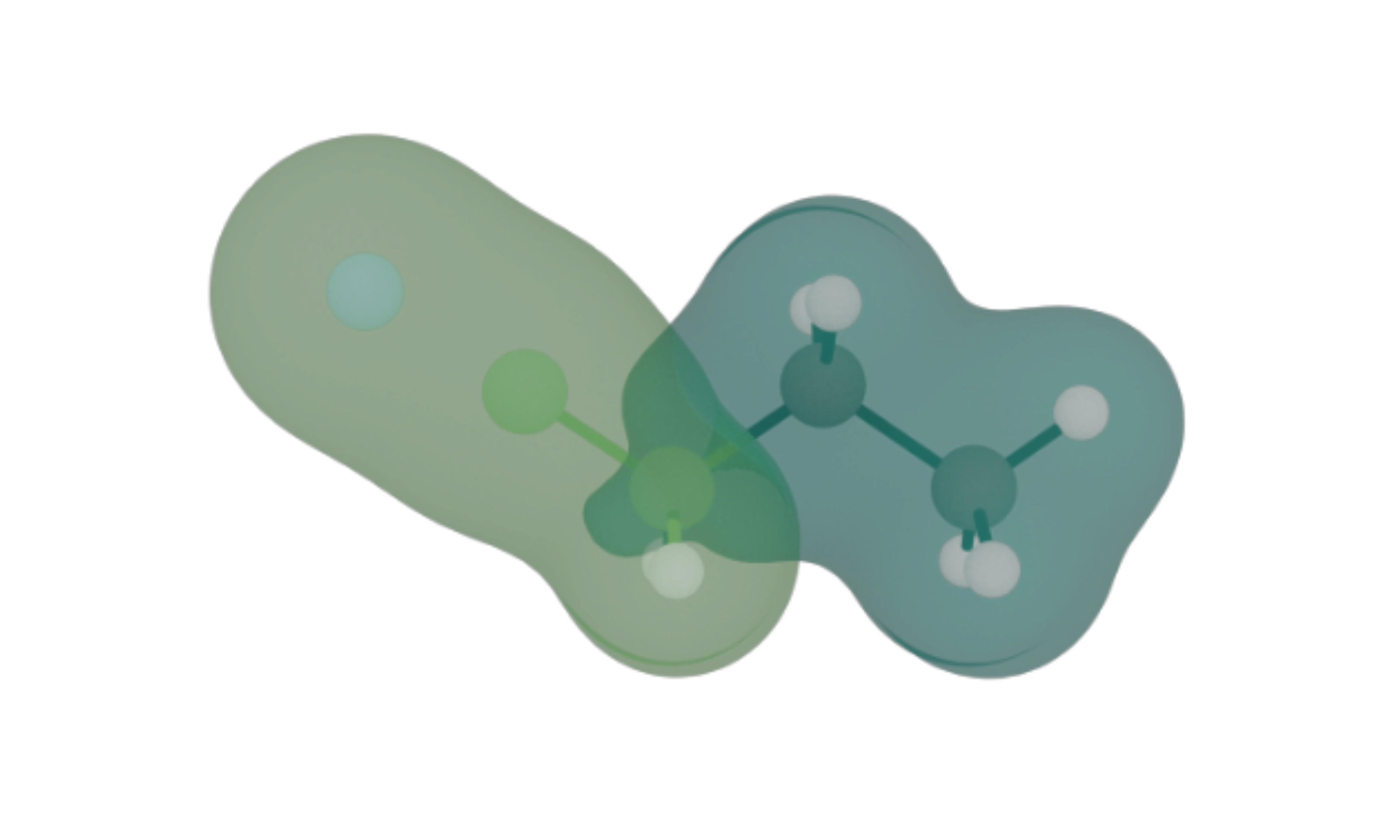
Simulating the dissociation is done by displacing the nitrogen atom of the nitrile group stepwise and calculating the system energy at each step. Using Qrunch it is straight-forward to implement the problem. The FAST-VQE algorithm is utilized to build a quantum circuit representing the wave-function, and quantum hardware is exploited for the task of adaptively selecting next operators to be added to iteratively converge towards the ground state. The energy evaluation is off-loaded to a classical computation.
In a series of computations performed on quantum computers on the IQM Resonance cloud, we have demonstrated how Qrunch enables progressive scaling of the problem to instances exploiting increasing qubit numbers. Calculations on 16 and 20 qubits were carried out on IQM's Sirius and Garnet devices, respectively, effectively pushing them to their limits in terms of qubit exploitation. Finally, the computation was scaled to 50 qubits on IQM Emerald.
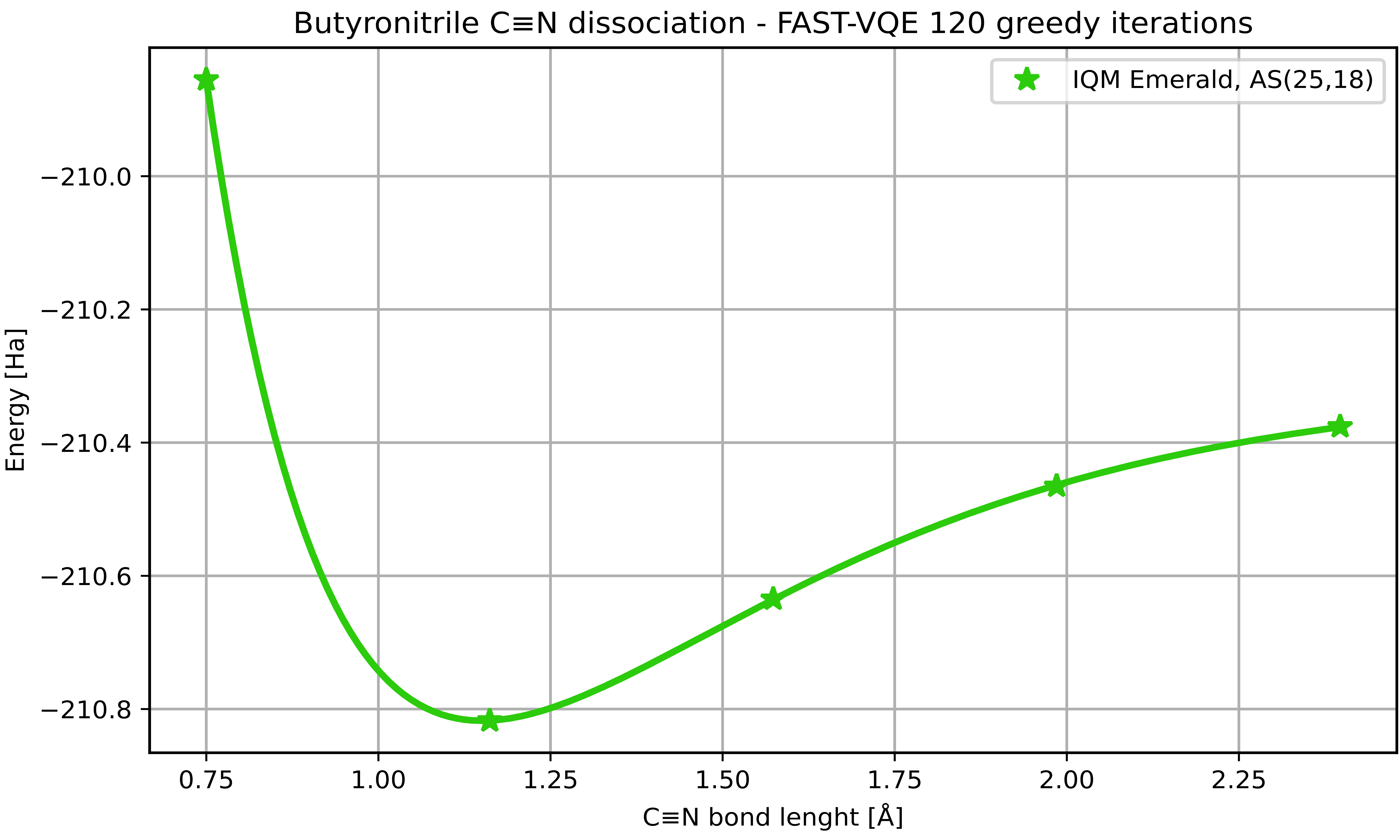
At the large active space deployed, AS(25,18), we cannot compare to a classical reference calculation as it is beyond reach of our classical CASCI capabilities. Also, the classical parameter optimization step in the VQE calculation starts to become a bottleneck. To enable scaling, this was mitigated by adopting a greedy approach where operator parameters were optimized one at a time and an approximate simulator was used for the VQE part.
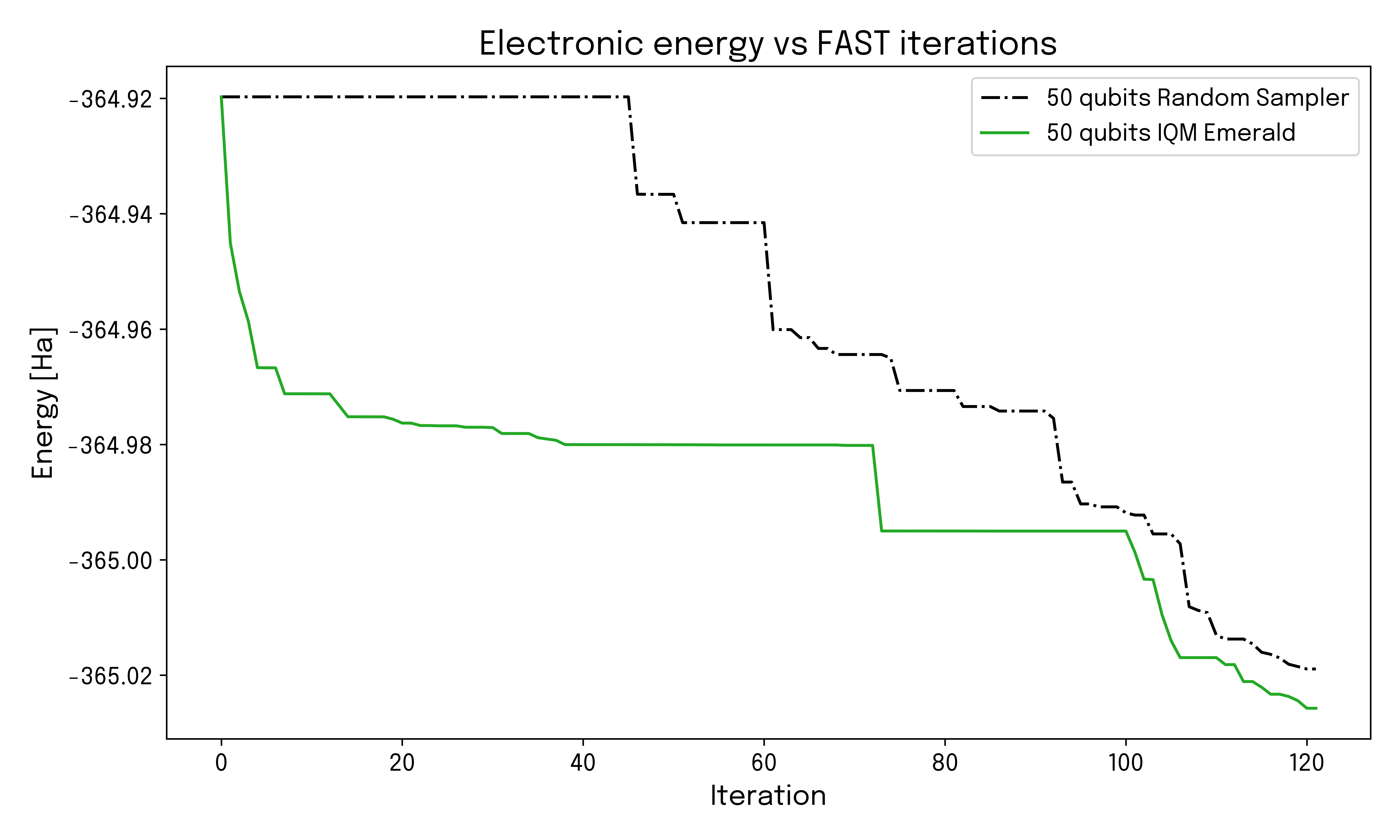
Key Results
The role of the quantum computer is to sample the increasingly complex quantum circuit and generate bitstrings that guide the selection of added operators. To assess the impact of noise on the quantum hardware performance and how much computational value is extracted from the qubits, we simulated the case where the hardware is completely dominated by noise and the output bitstring consequently completely random. Comparing this to the actual results from IQM Emerald shows that the actual quantum hardware delivers faster convergence, and the achieved accuracy is limited only by quantum hardware.
Conclusion
Using Qrunch, we simulated the dissociation of the nitrile group in butyronitrile — a promising electrolyte candidate for solar cells and future lithium-ion batteries — and demonstrated extreme scalability of the problem. Utilizing up to 50 qubits in computations executed on quantum hardware, we go beyond reach of exact classical methods and achieve an accuracy limited by quantum hardware only. This demonstrates the applicability and utility of current quantum computing solutions for problems of immediate relevance for the development of renewable energy technology.
Tutorial
A tutorial explaining how to set up the computations involved in this showcase is available here.
Understanding electrolyte degeneration through ionization energies
renewable energy
battery chemistry
Motivation
High-performance batteries are fundamental in modern vehicle manufacturing as well as in renewable energy production, where efficient energy storage is crucial for ensuring a reliable and stable supply. Efforts have been made towards the development of batteries with higher energy density, with the major bottleneck being that the higher voltages involved cause electrolyte degradation, especially at the cathode, where the specific reaction is an oxidation.
Ability to predict and model with high accuracy the resilience toward cathode oxidation for a range of electrolytes is needed. This would allow screening of potential new electrolytes prior to experimental testing, contributing to a more efficient development of future batteries. A particular point of interest in this context is the calculation of ionization energies of small molecules, that is, the reduction in the ground state energy of a molecule when it loses an electron.
The case
In this showcase, Qrunch is utilized to perform FAST-VQE calculations on quantum hardware to approximate the ionization energy of a simple molecule, namely H₂O. We aim to calculate the ionization energies of the H₂O molecule in the neutral and ionized regimes. As our molecular configuration, we consider the geometry of the H₂O molecule in its equilibrium state and assume the minimal basis set, STO-3G.

We also adopt an active space approach to limit the number of spatial orbitals and electrons used in the quantum algorithm. In the neutral configuration, we adopt AS(6,8), that is, 6 spatial orbitals and 8 electrons. This maps into 12 qubits and 8 excitations in a quantum circuit. In the ionized configuration, we correspondingly define the AS(6,7), where we have one less electron.
The ground state energies of the molecule are obtained by running the FAST-VQE algorithm. In this approach, we use samples obtained from quantum hardware or from a simulated state vector to iteratively select and add gates to the circuit. The gate pool includes single and double-excitation operators, which excite electrons from a reference state (the Hartree-Fock state). In all calculations, we fix 30 iterations for the adaptive run.
When sampling from quantum hardware, we are employing OQC's Toshiko device, which is a QPU with 32 qubits. Sampling for gate selection is performed using 1024 shots. The energy estimation is done classically, using Kvantify's Excitation Gate simulator, and gate parameters are globally optimized. Our criterion for convergence is reaching chemical accuracy, that is, an absolute error of approximately 1.6 mHa (or 1 kcal/mol).
Key Results
The energy error as a function of the number of iterations is displayed for both the state vector simulation and the quantum hardware run on OQC Toshiko, for both the neutral and ionized configurations. The reference values for the ground state energies used are obtained from a configuration interaction (CI) approach.
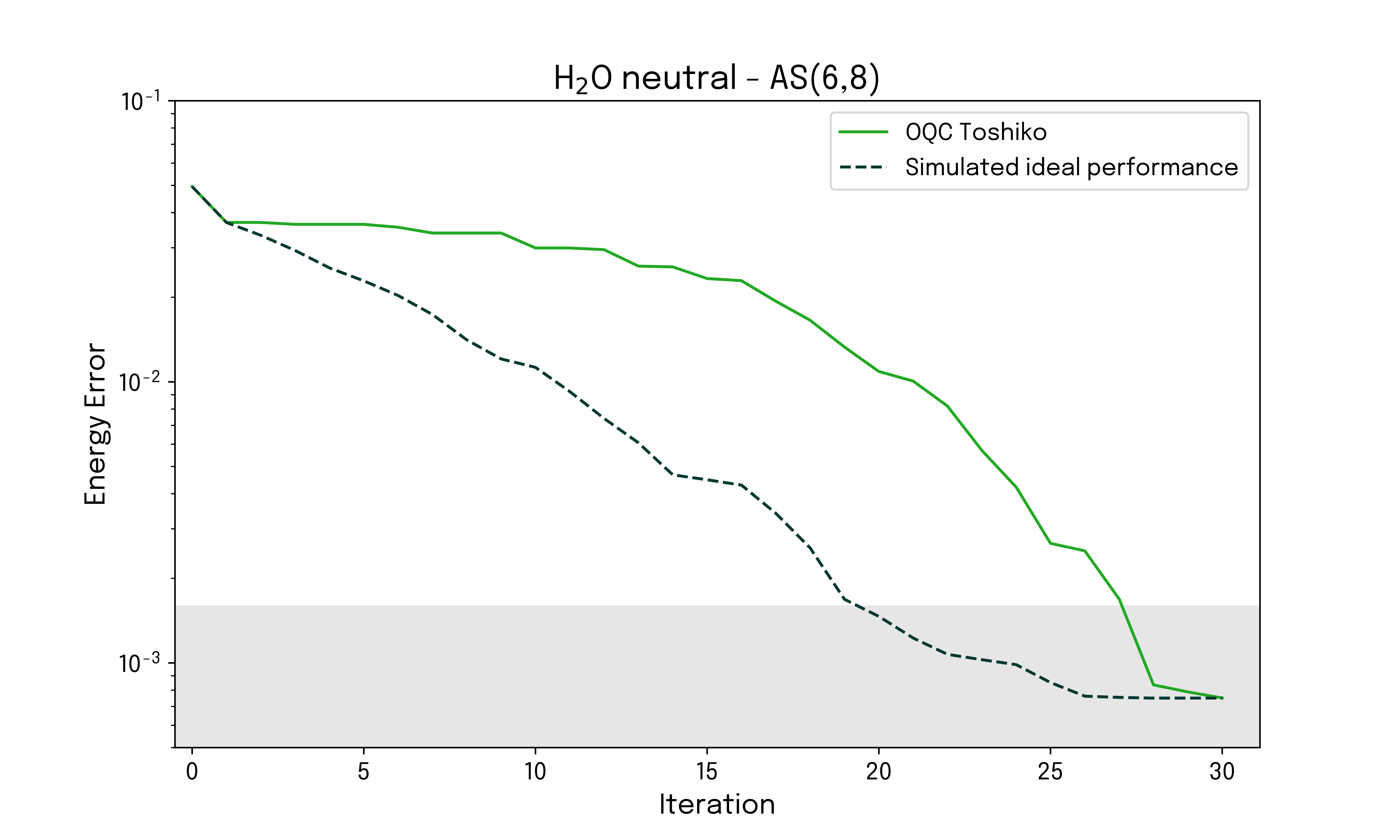
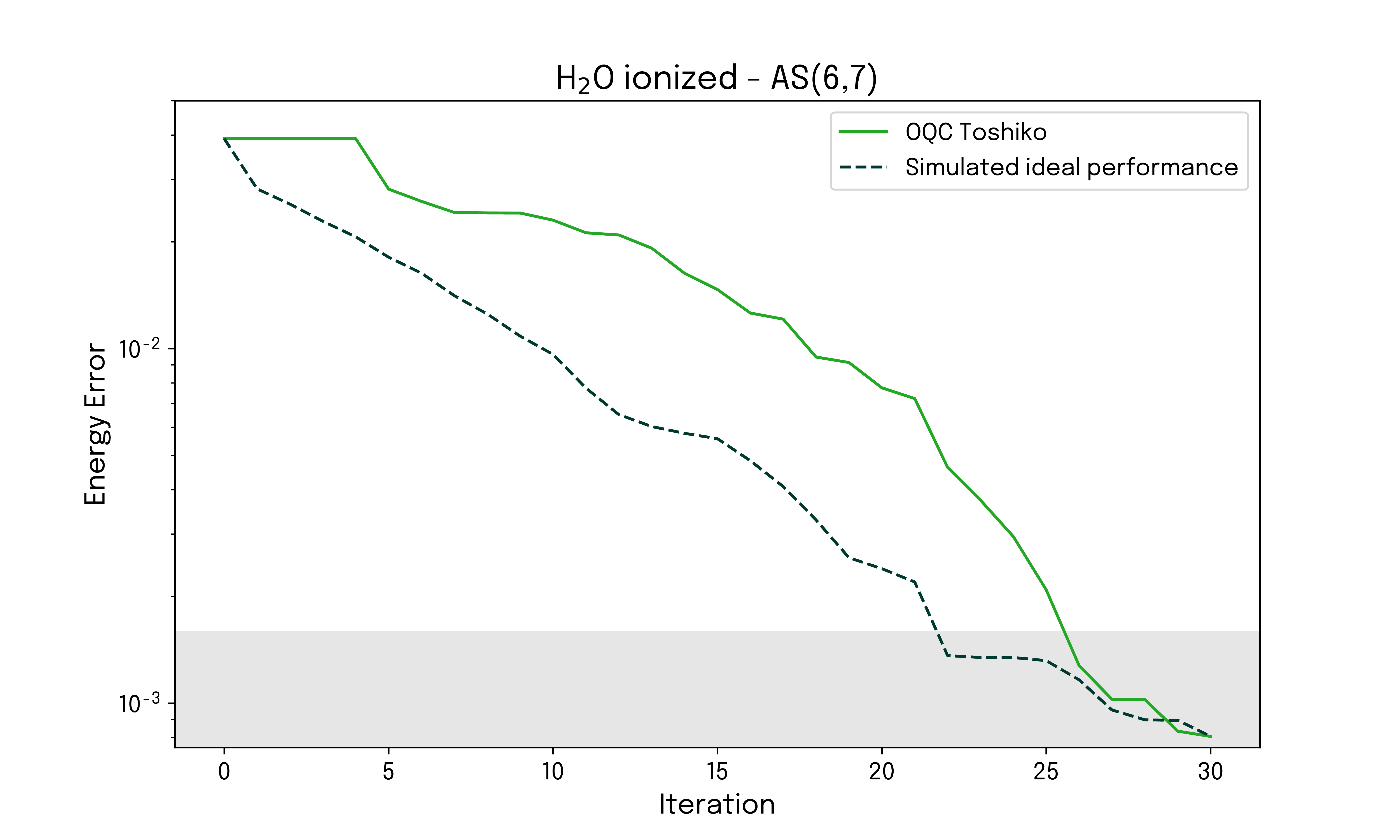
The targeted ionization energy corresponds to the difference between the neutral and ionized configuration energies, and the convergence of the ionization energy can readily be investigated. The result is given below.
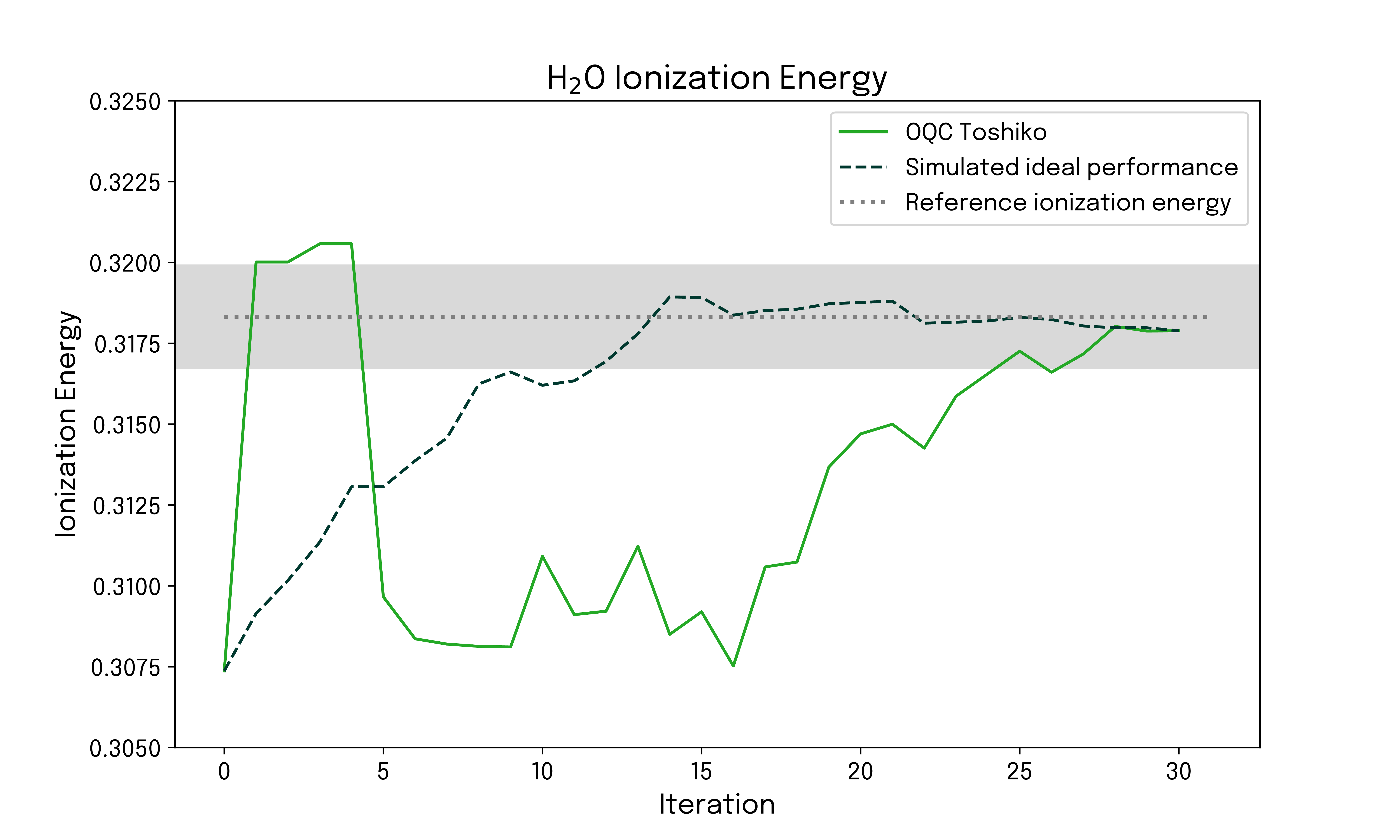
While in the above example, the energy estimation is always performed classically, we are able to find good convergence by selecting and applying gates to a reference state based on samples from quantum hardware. The quality of the results is validated by comparison to an ideal state vector simulation, and for both the neutral and ionized configuration, the energy error converges to below chemical accuracy within a few iterations. This behavior is also manifested in the derived ionization energy for the system.
Conclusion
Using Qrunch, we have run the FAST-VQE algorithm on 12 qubits of the OQC Toshiko quantum hardware and calculated the ionization energy of the H₂O molecule. This study showcases the possibility of employing current quantum hardware to address electrolyte degradation processes, which are commercially relevant for technology developments enabling increased electrification and transition to renewable energies.
Tutorial
A tutorial explaining how to set up the computations involved in this showcase is available here.